
Type I Disease (Most Common)
Idiopathic
Subacute bacterial endocarditis
Systemic lupus erythematosus
Hepatitis C ± cryoglobulinemia
Mixed cryoglobulinemia
Hepatitis B
Cancer: Lung, breast, and ovary (germinal)
Type II Disease (Dense Deposit Disease)
Idiopathic
C3 nephritic factor-associated
Partial lipodystrophy
Type III Disease
Idiopathic
Complement receptor deficiency
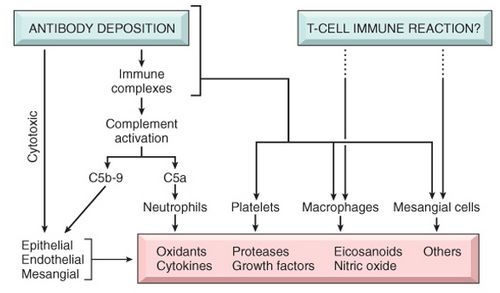
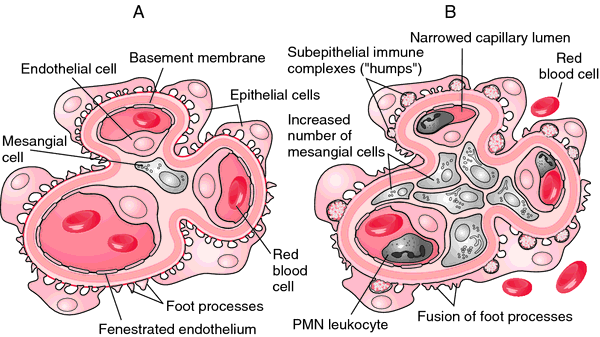
Type I MPGN, the most proliferative of the three types, shows mesangial proliferation with lobular segmentation on renal biopsy and mesangial interposition between the capillary basement membrane and endothelial cells, producing a double contour sometimes called tram-tracking.
Subendothelial deposits with low serum levels of C3 are typical, although 50% of patients have normal levels of C3 and occasional intra-mesangial deposits.
Low serum C3 and a dense thickening of the GBM containing ribbons of dense deposits and C3 characterize Type II MPGN, sometimes called dense deposit disease . Classically, the glomerular tuft has a lobular appearance; intramesangial deposits are rarely present and subendothelial deposits are generally absent.
Proliferation in Type III MPGN is less common than the other two types and is often focal; mesangial interposition is rare, and subepithelial deposits can occur along widened segments of the GBM that appear laminated and disrupted.
Type I MPGN is secondary to glomerular deposition of circulating immune complexes or their in situ formation.
Types II and III MPGN may be related to "nephritic factors," which are autoantibodies that stabilize C3 convertase and allow it to activate serum C3.
Patients with MPGN present with proteinuria, hematuria, and pyuria (30%), systemic symptoms of fatigue and malaise that are most common in children with Type I disease, or an acute nephritic picture with RPGN and a speedy deterioration in renal function in up to 25% of patients. Low serum C3 levels are common.
Fifty percent of patients with MPGN develop end-stage disease 10 years after diagnosis, and 90% have renal insufficiency after 20 years. Nephrotic syndrome, hypertension, and renal insufficiency all predict poor outcome. In the presence of proteinuria, treatment with inhibitors of the renin-angiotensin system is prudent. Evidence for treatment with dipyridamole, coumadin, or cyclophosphamide is not strongly established nor recommended. There is some evidence supporting the efficacy of treatment of primary MPGN with steroids, particularly in children.
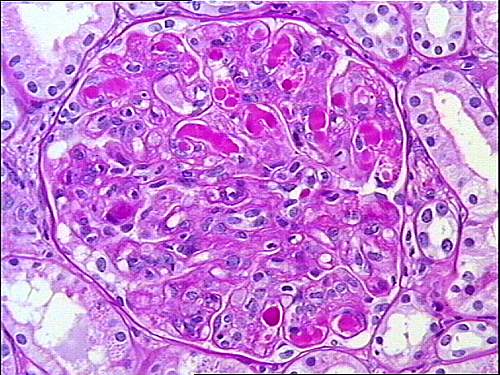 |
| membranoproliferative glomerulonephritis with huge intracapillary deposits (thrombi) totally filling the capillary lumina |
In secondary MPGN, treating the associated infection, autoimmune disease, or neoplasms is of demonstrated benefit. Although all primary renal diseases can recur over time in transplanted renal allografts, patients with MPGN are well known to be at risk for this adverse event.
No comments:
Post a Comment